




24小時(shí)服務(wù)熱線:
400-1846-454

激光產(chǎn)品全球其他市場(chǎng)準(zhǔn)入解決方案
激光產(chǎn)品中國(guó)市場(chǎng)準(zhǔn)入全方案
IEC 62471 光生物安全檢測(cè)與風(fēng)險(xiǎn)
EN 50689 消費(fèi)類激光產(chǎn)品專用安全
EN 60825-1 標(biāo)準(zhǔn)檢測(cè)與CE符合
FDA 21 CFR 1040.10標(biāo)準(zhǔn)



FDA 510(k)認(rèn)證是什么意思?
FDA 510(k)認(rèn)證是美國(guó)食品藥品監(jiān)督管理局(FDA)針對(duì)醫(yī)療器械上市前提交的一種預(yù)市通知(Premarket Notification)。根據(jù)《聯(lián)邦食品、藥品和化妝品法案》(FD&C Act)第510(k)條款,制造商在將某些醫(yī)療器械投放美國(guó)市場(chǎng)之前,必須向FDA證明其產(chǎn)品與已合法上市的“謂詞設(shè)備”(Predicate Device)在安全性和有效性方面“實(shí)質(zhì)等同”(Substantially Equivalent, SE)。
510(k)認(rèn)證的核心目的是確保新醫(yī)療器械不會(huì)對(duì)患者或使用者造成額外的風(fēng)險(xiǎn),同時(shí)能夠達(dá)到與現(xiàn)有產(chǎn)品相同的預(yù)期用途。與更嚴(yán)格的PMA(Premarket Approval)不同,510(k)路徑適用于中低風(fēng)險(xiǎn)的II類醫(yī)療器械(部分I類器械可豁免)。
認(rèn)證詳解:定義、適用范圍、要求與流程(圖1)")
FDA 510(k)認(rèn)證適用哪些產(chǎn)品?
510(k)認(rèn)證主要適用于II類醫(yī)療器械,部分I類器械(如帶有特殊功能的)也可能需要提交。常見需要510(k)認(rèn)證的產(chǎn)品包括:
醫(yī)療激光產(chǎn)品(如手術(shù)激光、治療激光、診斷激光設(shè)備)
影像診斷設(shè)備(如超聲、X射線設(shè)備)
體外診斷設(shè)備(如血糖儀、某些試劑)
骨科植入物(如人工關(guān)節(jié)、骨釘)
監(jiān)護(hù)設(shè)備(如心電圖機(jī)、血壓監(jiān)測(cè)儀)
牙科設(shè)備(如牙科激光、種植體)
值得注意的是,并非所有醫(yī)療器械都需要510(k)認(rèn)證。例如,部分低風(fēng)險(xiǎn)的I類器械(如普通手術(shù)器械)可能豁免510(k),而高風(fēng)險(xiǎn)的III類器械(如心臟起搏器)則需走PMA路徑。
FDA 510(k)認(rèn)證的要求
要獲得510(k)認(rèn)證,制造商需滿足以下核心要求:
(1)確定產(chǎn)品分類和監(jiān)管路徑
查詢FDA產(chǎn)品分類數(shù)據(jù)庫,確認(rèn)產(chǎn)品是否屬于II類器械,并確定適用的產(chǎn)品代碼(Product Code)和法規(guī)編號(hào)(21 CFR部分)。
(2)選擇正確的謂詞設(shè)備
需選擇至少一個(gè)已合法上市的美國(guó)醫(yī)療器械作為對(duì)比對(duì)象,證明新設(shè)備在預(yù)期用途、技術(shù)特性、安全性和性能方面與之實(shí)質(zhì)等同。
(3)提供科學(xué)合理的測(cè)試數(shù)據(jù)
包括生物相容性測(cè)試(ISO 10993)、電氣安全測(cè)試(IEC 60601)、電磁兼容性測(cè)試(EMC)、性能測(cè)試等。
對(duì)于醫(yī)療激光產(chǎn)品,需符合FDA 21 CFR 1040.10標(biāo)準(zhǔn),確保激光輻射安全。
(4)符合質(zhì)量管理體系(QMS)要求
制造商需建立符合21 CFR 820(QSR)的質(zhì)量管理體系,確保產(chǎn)品設(shè)計(jì)、生產(chǎn)、包裝和標(biāo)簽符合FDA要求。
FDA 510(k)認(rèn)證流程
510(k)認(rèn)證流程通常包括以下步驟:
產(chǎn)品分類確認(rèn):確定產(chǎn)品屬于I、II或III類,并確認(rèn)是否需要510(k)。
選擇謂詞設(shè)備:在FDA數(shù)據(jù)庫(如510(k) Premarket Notification或De Novo數(shù)據(jù)庫)中尋找合適的對(duì)比設(shè)備。
準(zhǔn)備技術(shù)文件:包括產(chǎn)品描述、性能測(cè)試報(bào)告、生物相容性數(shù)據(jù)、軟件驗(yàn)證(如適用)、標(biāo)簽和說明書等。
提交510(k)申請(qǐng):通過FDA的電子提交門戶(eSubmitter或CDRH Portal)遞交申請(qǐng)。
FDA審核:FDA通常在90天內(nèi)完成審核,可能要求補(bǔ)充數(shù)據(jù)(RTA)。
獲得許可:如審核通過,F(xiàn)DA會(huì)發(fā)放“Substantially Equivalent”(SE)決定函,允許產(chǎn)品在美國(guó)上市。
FDA 510(k)認(rèn)證所需資料
510(k)提交的核心文件包括:
文件類型 | 具體內(nèi)容 |
行政信息 | 企業(yè)信息、產(chǎn)品名稱、型號(hào)、分類代碼等 |
設(shè)備描述 | 技術(shù)規(guī)格、工作原理、預(yù)期用途、與謂詞設(shè)備的對(duì)比分析 |
性能測(cè)試報(bào)告 | 電氣安全、EMC、生物相容性、軟件驗(yàn)證(如適用) |
激光安全數(shù)據(jù)(醫(yī)療激光產(chǎn)品) | 符合FDA 21 CFR 1040.10的輻射安全測(cè)試 |
標(biāo)簽和說明書 | 英文標(biāo)簽、使用說明、警告信息 |
質(zhì)量管理體系 | 符合21 CFR 820的QMS聲明 |
FDA 510(k)認(rèn)證周期
標(biāo)準(zhǔn)510(k):通常需幾個(gè)月(從提交到獲批)。
特殊510(k)(適用于輕微修改):可能縮短至1-2個(gè)月。
De Novo 510(k)(無謂詞設(shè)備的新類別):周期會(huì)更長(zhǎng),需要提前預(yù)留充足時(shí)間。
實(shí)際周期受FDA審核速度、數(shù)據(jù)完整性及企業(yè)響應(yīng)時(shí)間影響,具體周期可以咨詢專業(yè)機(jī)構(gòu)。
FDA 510(k)認(rèn)證注意事項(xiàng)
確保數(shù)據(jù)完整性:測(cè)試報(bào)告必須由專業(yè)實(shí)驗(yàn)室出具,避免因數(shù)據(jù)不全導(dǎo)致審核延遲。
正確選擇謂詞設(shè)備:如選擇錯(cuò)誤的對(duì)比設(shè)備,可能導(dǎo)致FDA拒絕“實(shí)質(zhì)等同”判定。
標(biāo)簽和說明書合規(guī):所有標(biāo)簽必須使用英文,并符合FDA的UDI(唯一設(shè)備標(biāo)識(shí))要求。
持續(xù)合規(guī):獲批后仍需遵守FDA的上市后監(jiān)管要求,如不良事件報(bào)告(MDR)。
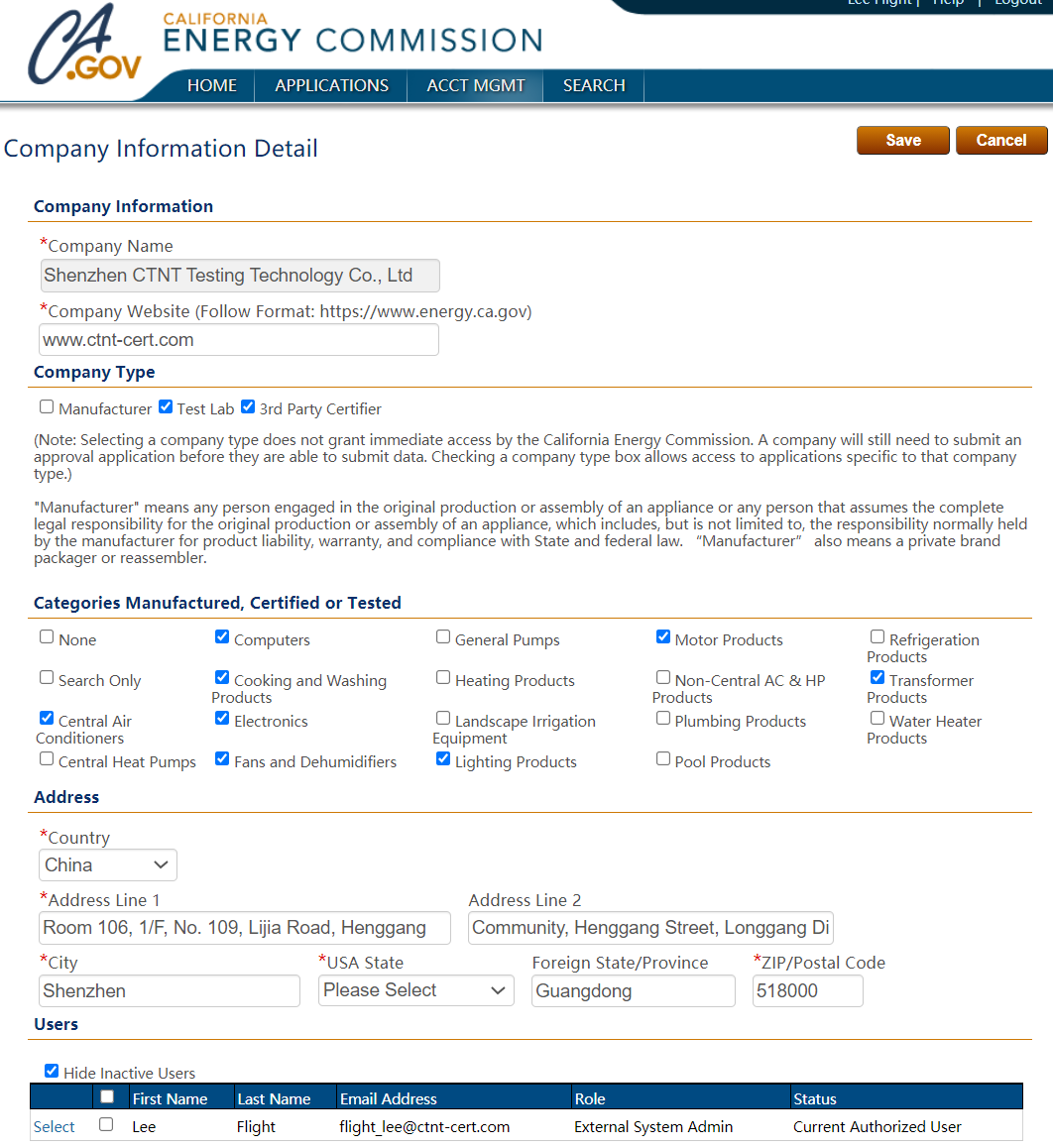
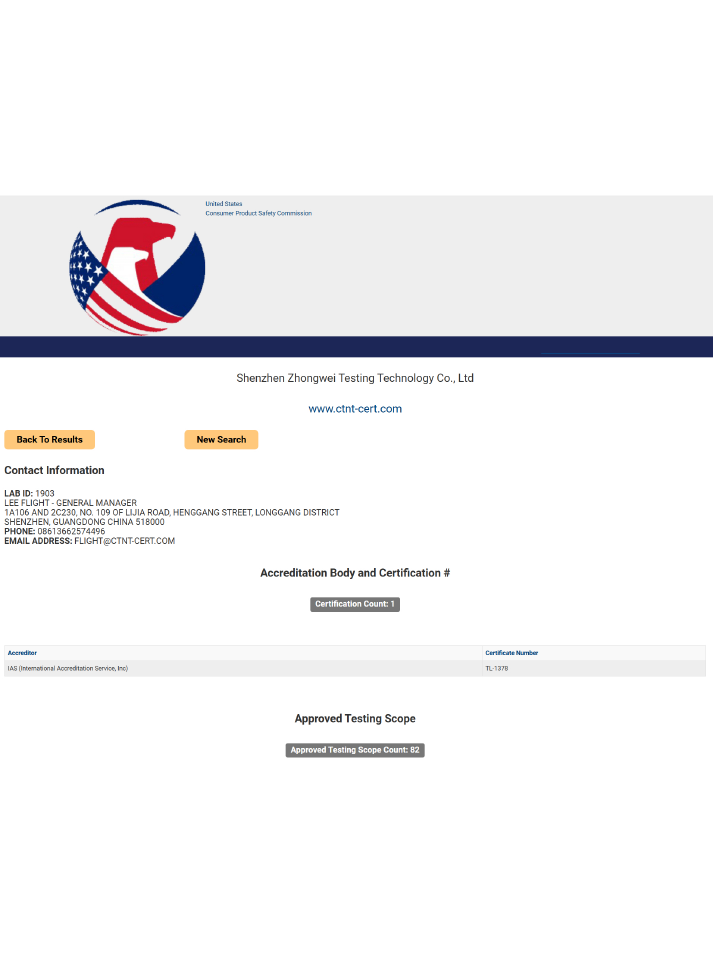
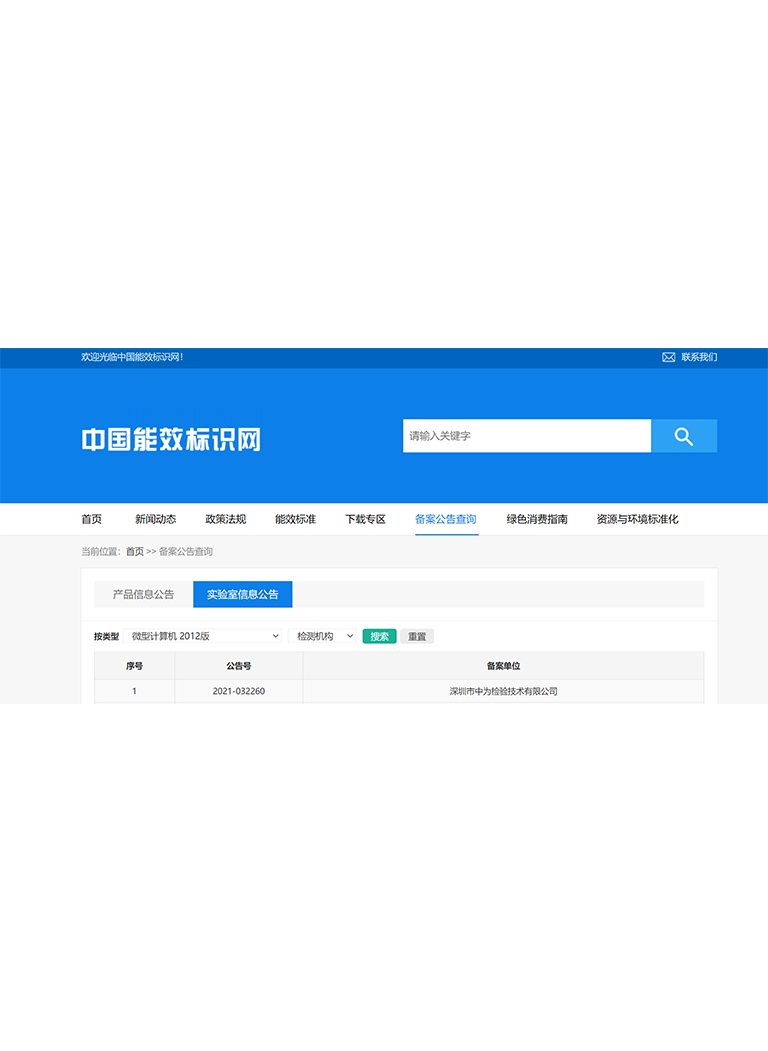
深圳中為檢驗(yàn):專業(yè)激光產(chǎn)品FDA 510(k)檢測(cè)服務(wù)
深圳市中為檢驗(yàn)技術(shù)有限公司是一家專業(yè)的激光產(chǎn)品檢測(cè)和認(rèn)證機(jī)構(gòu),專注于醫(yī)療激光、工業(yè)激光、消費(fèi)激光等產(chǎn)品的FDA認(rèn)證服務(wù)。
我們的服務(wù)范圍包括:
醫(yī)療激光產(chǎn)品FDA 510(k)檢測(cè)(符合21 CFR 1040.10標(biāo)準(zhǔn))
工業(yè)激光產(chǎn)品FDA認(rèn)證
消費(fèi)激光產(chǎn)品FDA認(rèn)證
測(cè)試測(cè)量激光產(chǎn)品FDA認(rèn)證
表演和顯示類激光產(chǎn)品FDA認(rèn)證
激光美容儀FDA認(rèn)證
激光雷達(dá)FDA認(rèn)證
我們擁有FDA認(rèn)可的測(cè)試實(shí)驗(yàn)室,可提供完整的510(k)技術(shù)文件支持,幫助企業(yè)高效完成認(rèn)證并進(jìn)入美國(guó)市場(chǎng)。
為什么選擇我們?
專業(yè)團(tuán)隊(duì):10年以上FDA認(rèn)證經(jīng)驗(yàn),熟悉最新法規(guī)要求(截至2025年)。
一站式服務(wù):從測(cè)試到文件編寫、提交,全程支持。
快速響應(yīng):優(yōu)化流程,縮短認(rèn)證周期。
如果您有醫(yī)療激光產(chǎn)品或其他激光設(shè)備需要出口美國(guó),歡迎聯(lián)系深圳中為檢驗(yàn),我們將為您提供專業(yè)的FDA 510(k)認(rèn)證服務(wù)!






返回頂部

電話:400-1846-454

地址:深圳市龍崗區(qū)橫崗街道橫崗社區(qū)力嘉路109號(hào)
 網(wǎng)站備案:粵ICP備2020126073號(hào)
網(wǎng)站備案:粵ICP備2020126073號(hào)
 公安網(wǎng)備案:公安網(wǎng)備44030602005252號(hào)
網(wǎng)絡(luò)違法信息舉報(bào)平臺(tái)
公安網(wǎng)備案:公安網(wǎng)備44030602005252號(hào)
網(wǎng)絡(luò)違法信息舉報(bào)平臺(tái)
 當(dāng)前位置:
當(dāng)前位置: 







